Diagnosis of late onset neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS; motor neuron disease, MND) or progressive supranuclear palsy (PSP) relies on a series of clinical tests to exclude other conditions and can lead to a delay in diagnosis of many months. It is also difficult to predict the speed in which the disease will progress with any certainty. Therefore, finding a biomarker (an indicator of the disease) in blood could not only decrease this delay, but also predict prognosis and aid the development of new treatments. We are working on identifying a group of molecules called non-coding RNA (ncRNA) in the blood of ALS or PSP patients as potential biomarkers. In the case of ALS, this work has led to the identification of seven ncRNAs that allow us to predict if a sample is from an ALS patient or not (Joilin et al, Brain Comm, 2020). In our efforts for identifying a biomarker signature of the disease we are seeking for ncRNA biomarkers that change with the disease over time so that we can both predict and track disease progression. This is done with the use of samples collected from ALS patients over the course of their disease and will allow us to understand key differences between patients and aid in treatment development, leading to improved outcomes for people affected by ALS.
We are also investigating the importance of two ALS-associated proteins known as FUS and TDP-43 in response to genotoxic agents. We have shown that FUS is a component of the cellular response to DNA damage, and that defects in this response may contribute to ALS (Rulten et al, Nucleic Acids Res, 2014). In addition, through funding from the Motor Neurone Disease Association (MNDA), we are identifying non-coding RNAs, including microRNAs, as diagnostic and prognostic biomarkers for ALS.
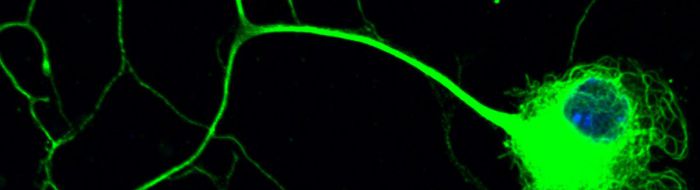
Motor neurons are amongst the most specialised and largest cells in the body so that the axon of a spinal motor neuron which innervates a muscle in the foot can be about a meter long in humans. These neurons are dependent on molecular motors, such as cytoplasmic dynein, and their cytoskeletal tracks for axonal transport and guidance of the axons towards their targets for the establishment and maintenance of synaptic connections.
As such, we have shown that cytoplasmic dynein-mediated axonal transport is impaired in the motor neurons of SOD1G93A transgenic mouse model of amyotrophic lateral sclerosis (ALS), an adult-onset and lethal form of motor neuron disease, even in the embryonic stages of development [Kieran et al, J Cell Biol, 2005]. In addition, in a mouse model of a childhood form of motor neuron disease known as spinal muscular atrophy, lower extremity, dominant (SMA-LED), we have shown that a mutation in the heavy chain subunit of cytoplasmic dynein causes neurodegeneration as a result of aberrant interaction of mutant dynein with its adaptor protein dynactin [Hafezparast et al, Science, 2003; Deng et al, J Biol Chem, 2010]. Subsequently, we have shown that this defect leads to impaired retrograde axonal transport of signaling endosomes [Garrett et al, Brian, 2014].
Children affected with SMA-LED exhibit delayed motor milestones and abnormal gait with lower limb muscle wasting, which are frequently associated with cognitive impairment. Moreover, fragmentation of the Golgi apparatus is commonly observed in both ALS and SMA-LED. Our research is currently focused on elucidating (1) the molecular mechanisms of motor neuron degeneration and cognitive impairment caused by mutations in the components of cytoplasmic dynein and its adaptor proteins; (2) the link between mutations in cytoplasmic dynein and the observed fragmentation of the Golgi apparatus in motor neuron disease.


